

- Name
- Rachel Butch
- rbutch1@jhmi.edu
- Office phone
- 410-955-8665
- Name
- Vanessa Wasta
- wasta@jhmi.edu
- Office phone
- 410-955-8236
In experiments performed in mice, Johns Hopkins researchers report they have identified the cascade of cell death events leading to the physical and intellectual degeneration associated with Parkinson's disease.
Results of the study, published today in Science, suggest promising new targets for drugs that could interrupt Parkinson's disease progression.
"Nailing down how cells die in this disease increases our hope that someday we will be able to treat and possibly cure Parkinson's disease," says Ted Dawson, director of the Institute for Cell Engineering and professor of neurology at the Johns Hopkins University School of Medicine.
The study focused on Parthanatos—a specialized "programmed" pathway toward cellular death named for Thanatos, the ancient Greek personification of death—as a key driver of nerve cell degradation that is a marker of Parkinson's disease. It is distinct from other known forms of programmed cell death such as apoptosis (a normal part of growth and development) and necroptosis (generally, cell death due to disease or injury).
The first step down Parthanatos in Parkinson's disease is the accumulation of misfolded proteins in brain neurons. These proteins, known as alpha synuclein, have long been linked to Parkinson's disease progression, but it was unclear how they specifically affected brain cells.
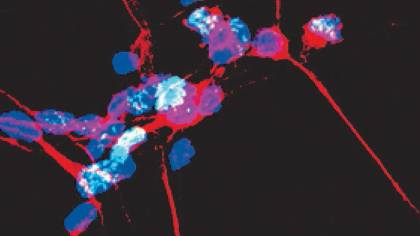
Image caption: Dopamine neurons (pictured) are the primary cell type targeted by Parthanatos in Parkinson’s disease
Image credit: The Dawson Lab
To find out more about which "road" to cell death Parkinson's cells travel, the research team treated lab-grown mouse brain cells with preformed clumps of alpha synuclein and observed their response over the course of 14 days. As the brain cells began to die off, the researchers observed that they had "turned on" a protein called PARP1, a gateway to cell death via Parthanatos.
They then tested whether blocking PARP1 could rescue the cells from certain death. In an additional experiment, the researchers again added alpha synuclein clumps to healthy mouse brain cells and then treated the cells with one of three drugs that block PARP1's function: veliparib, rucaparib, or talazoparib, all currently used by oncologists to treat breast and ovarian cancers. The researchers found that the cells treated with these drugs were protected from death at 14 days.
To test this principle in living mammals, the research team injected alpha synuclein clumps into the brains of normal mice and mice genetically engineered to lack the PARP gene. The researchers found that the normal mice began to show muscle weakness, loss of coordination, and decreased movement, as seen in tests of the mice's grip strength and the ability to climb down a vertical pole, three months after treatment. However, both the mice lacking PARP and normal mice treated with PARP blockers showed no decline.
"Showing that blocking this key step in the Parthanatos pathway protected the cells against death is evidence that Parkinson's disease kills cells through this mechanism," says Tae-In Kam, lead author on the study and postdoctoral fellow in the Institute for Cell Engineering at the Johns Hopkins University School of Medicine.
Past studies showed that PARP causes neurons to create a sugar called PAR, which binds to alpha synuclein and increases the rate at which the alpha synuclein proteins clump together. Kam wondered whether the increase in PARP1 he observed in Parkinson's cells could be causing a similar effect.
To test this hypothesis, the researchers added the PAR sugar along with the preformed alpha synuclein clumps to lab-grown mouse brain cells. They found that the combination of the sugar and alpha synuclein formed a different, more neurotoxic strain of the alpha synuclein clump. The cells treated with this combination died off 25 times faster than their counterparts receiving alpha synuclein alone.
To validate this observation, the research team repeated the experiment in normal mice. The researchers administered the preformed alpha synuclein clumps or the more toxic PAR/alpha synuclein combination to the mice's brains and again observed them for six months. The mice who received only alpha synuclein clumps began to show signs of degeneration six months after treatment. However, the mice that received the combination treatment experienced degeneration twice as fast, exhibiting significant degeneration at just three months.
"The PAR/alpha synuclein combination is not only faster at killing neurons, it's a more potent toxin," Kam says.
To gauge whether this mechanism could be in play in human Parkinson's disease, the researchers collected cerebrospinal fluid from 21 Parkinson's disease patients in various stages of the disease, and fluid samples from 33 healthy people. The team then measured the amount of PAR in each sample. What they found was that there was approximately twice the amount of PAR in the samples from people with Parkinson's disease.
"Additionally, one out of four of our samples showed a correlation between PAR concentration and the disease's progression," Kam says.
The researchers emphasized that much more research needs to be done before their findings can be applied to humans, but if further experiments support their results, the researchers hope to work on clinical trials with drug companies that currently produce drugs targeting PARP to test the ability of these drugs to slow, or even stop, Parkinson's disease in humans.
"If PARP inhibitors work in human Parkinson's disease patients as they have in mice, they could be protective of cells already affected by Parkinson's disease, but also slow the transmission of these harmful proteins to new cells," says Valina Dawson, professor of neurology at the Johns Hopkins University School of Medicine.
Posted in Health
Tagged parkinson's disease, cell engineering
